What is Congenital Diaphragmatic Hernia?
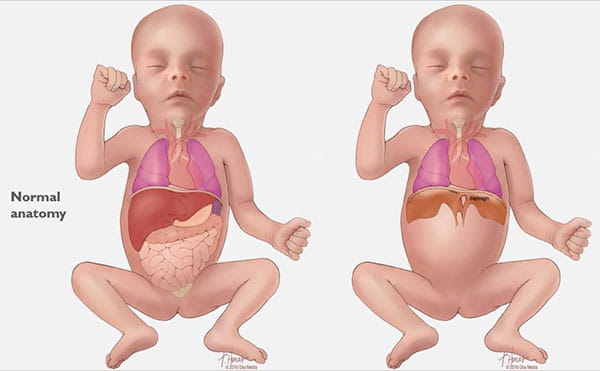
Congenital diaphragmatic hernia (CDH) is a defect in an unborn baby’s diaphragm, the muscle that divides the chest cavity and abdominal cavity.
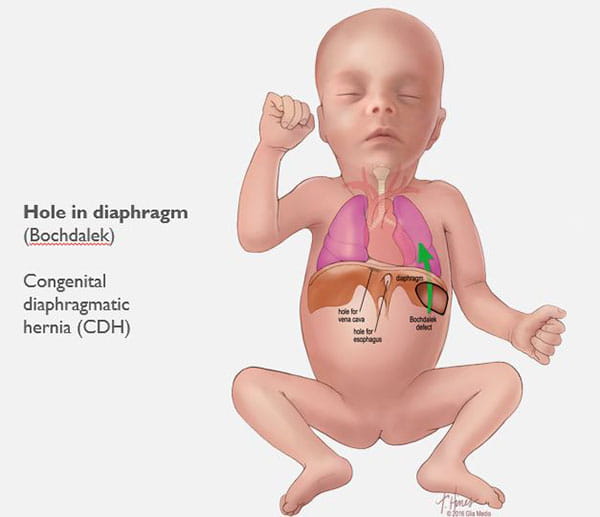
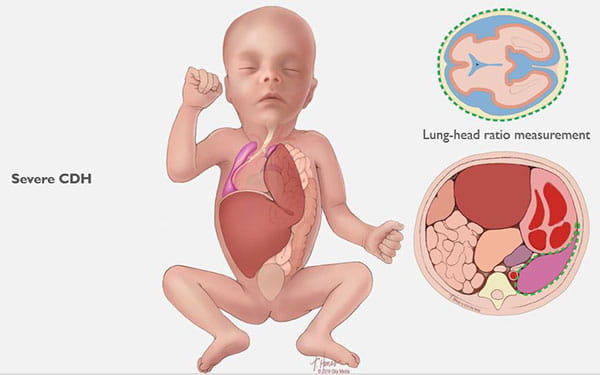
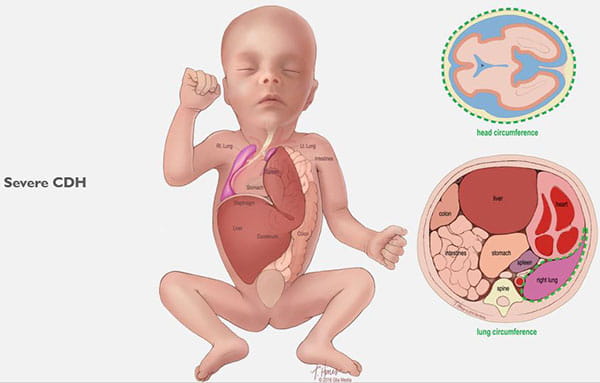
CDH occurs when the diaphragm does not close the right way during the baby’s development and abdominal organs push (“herniate”) through the defect into the chest cavity.
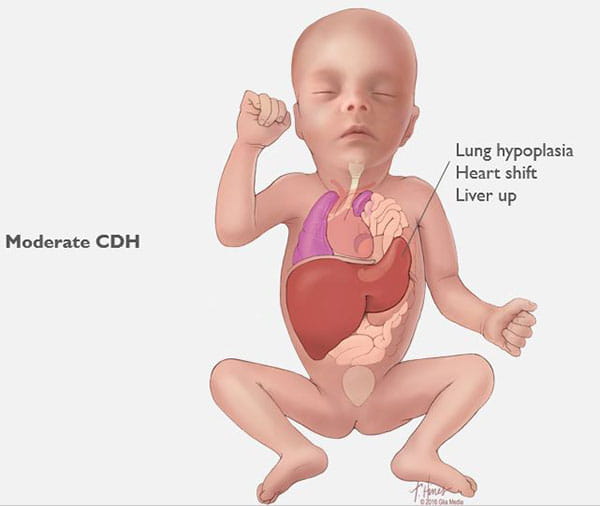
This can occur on the left or right side, but left is more common. The organs that come up into the chest can press on the lungs and can result in a condition in which the lungs remain small and underdeveloped, also called pulmonary hypoplasia. Some babies with CDH also develop conditions affecting their heart, liver, intestines and nervous system development.
CDH is not usually inherited, and its cause is not known. CDH occurs in about one in 2,200 live births and is usually seen during a routine prenatal ultrasound in the first or second trimester.
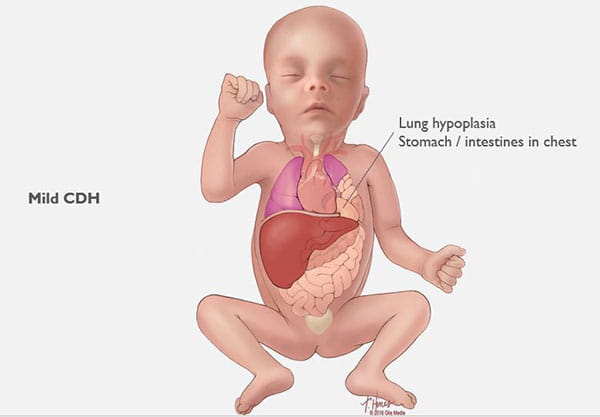
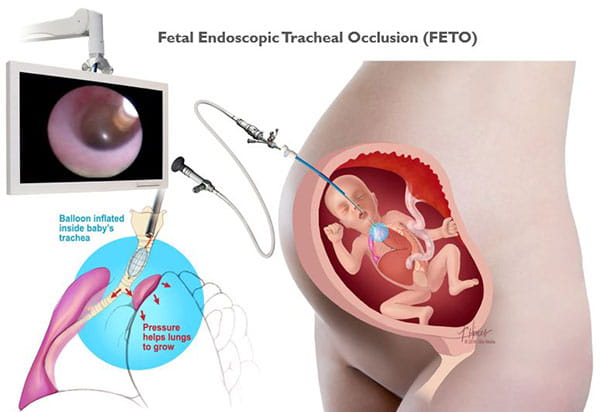
Congenital diaphragmatic hernias can range from mild to severe. In the best cases, infants do very well with routine prenatal and neonatal care, and may not need emergency surgery after birth. Lungs in these babies with a mild form of CDH will still be smaller than normal, but they have the chance to grow and adapt for many years. Many of these children can lead normal, active lives without long-term problems.
Babies with severe CDH will struggle after birth. Some will not survive. Others are saved through intensive medical care. However, these babies often have long-term health issues with breathing, feeding, growth, hearing and development.