
Cracking the Genetic Code of Malignant Peripheral Nerve Sheath Tumors
|
Top Breakthrough Discovery | Published February 2018 in Cancer Cell |

Q. Richard Lu, PhD and Lai Man Wu, PhD
A far-reaching effort to genetically profile a collection of malignant peripheral nerve sheath tumors (MPNSTs) has identified a potential therapy that shows initial success at inhibiting tumor growth in human cells and mouse models.
MPNSTs are rare, aggressive tumors that occur in about 0.001 percent of the general population, according to the National Institutes of Health. They often resist radiation and chemotherapy, and are known for high relapse rates and poor prognosis.
The breakthrough study was led by Q. Richard Lu, PhD, Scientific Director of the Brain Tumor Center, and Lai Man Natalie Wu, PhD, a research fellow in the Lu Lab. The project involved collaborators based in Boston, Dallas, Houston, St. Louis and Germany—and required obtaining robotic equipment to complete the analysis.
“We uncovered potential therapeutic targets that we did not expect for these untreatable tumors,” Lu says. “But our findings also need further study before knowing whether they will be relevant to patient treatment in the clinic.”
A mechanism of runaway cell growth
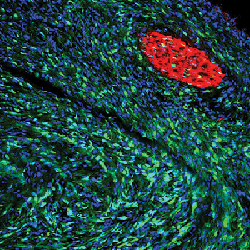
A sciatic nerve tumor from a Lats1/2-def mouse shows a surge in Sox10+ Schwann cell-derived tumor cells (green).
MPNSTs develop in Schwann cells, which form the myelin sheath that insulates peripheral nerves. About half of MPNSTs are linked to mutations of the NF1 gene, but the other half have no known genetic origins.
Normally, NF1 helps control a balanced rate of cell growth. When it mutates, it can cause neurofibromatosis 1, which typically leads to formation of brown skin spots and benign tumors along peripheral nerves. In some cases, NF1 mutation leads to runaway cell growth, creating large and medically problematic plexiform tumors. Some of these tumors can become MPNSTs upon acquiring additional gene mutations.
Digging deeper, Lu and colleagues detected an important role played by the gene Lats1/2, which normally suppresses cancer. When the gene’s expression is blocked, cells begin to rapidly multiply and become cancerous. Loss of Lats1/2 also causes other genes in the HIPPO signaling pathway (which controls tissue growth) to become hyperactive.
The team learned that two proteins associated with these hyperactive genes—TAZ and YAP—go on to interact with the transcription factor TEAD1 to activate molecular cancer programs that form MPNSTs.
Disrupting a deadly cascade
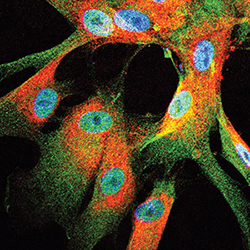
A micrograph highlighting the presence of TAZ/YAP (green) in human malignant peripheral nerve sheath tumor cells.
The team used a small interfering RNA (siRNA) approach to disrupt overactive TAZ-YAP activity in tumor Schwann cells lacking Lats1/2. They also achieved pharmacological inhibition of TAZ-YAP using the compounds verteporfin and dobutamine. Moreover, suppressing TAZ-YAP function in mice bred to lack Lats1/2 reduced tumor numbers.
These efforts also blocked signaling from platelet-derived growth factor receptor (PDGF), which supports tissue growth. Combined, these steps reduced the size and number of MPNSTs in the mice.
Further study revealed that human MPNSTs also exhibit elevated HIPPO-TAZ-YAP expression. A similar siRNA knockdown approach also inhibited MPNST growth in human cells and caused tumor cell death.
This suggested that several known medications may be useful as therapies against nerve sheath tumors.
Search continues for best combination therapy
However, the team learned that monotherapies aimed at suppressing TAZ-YAP activity or PDGF signaling alone were not effective.
The team explored two TAZ-YAP blockers, verteporfin and dobutamine, and two PDGF receptor inhibitors, imatinib and sorafenib. While some of the drugs showed tumor-reducing effects in vitro, none of them reversed disease in vivo.
After trying various drug combinations at various dosages, the team reported the strongest in vivo reductions in tumor size and weight from a combination of verteporfin and sorafenib.
“However, in our in vivo treatment studies, we observed a delay of tumor growth rather than tumor regression, suggesting that the drugs tested exerted cytostatic instead of cytotoxic effects,” Lu says. “Further investigation into more druggable targets and combination therapeutic options is warranted for effective inhibition of tumor growth.”
Since publishing this paper, Lu and his colleagues have presented their findings at a neurofibromatosis conference in Paris, and at the Cold Spring Harbor Mechanisms and Models of Cancer meeting in New York. They also have performed a small molecule compound screen using the tumor cell lines derived from their animal models. Results of that work will be published in an upcoming paper.
“We are testing the efficacy of HIPPO-TAZ-YAP inhibitors in combination with other oncogenic pathway inhibitors,” Lu says. “If successful, this could lead to a better treatment of MPNSTs.”
Citation
Wu LMN, Deng Y, Wang J, Zhao C, Wang J, Rao R, Xu L, Zhou W, Choi K, Rizvi TA, Remke M, Rubin JB, Johnson RL, Carroll TJ, Stemmer-Rachamimov AO, Wu J, Zheng Y, Xin M, Ratner N, Lu QR. Programming of Schwann Cells by Lats1/2-TAZ/YAP Signaling Drives Malignant Peripheral Nerve Sheath Tumorigenesis. Cancer Cell. 2018 Feb 12;33(2):292-308.e297.


