Taming a Fatal Blood Cancer
Taming a Fatal Blood Cancer
Preclinical Study Points to Potential Treatment Targets for Aggressive Pediatric Leukemia
Wednesday, July 11, 2018
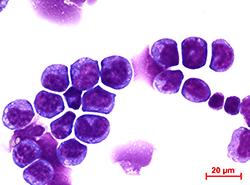
This microscopic image shows MLL-AF9 leukemia cells. Scientists who conducted extensive genetic and biochemical tests on the cells report in the Journal of Experimental Medicine identifying a potential treatment target for the aggressive, high-relapse leukemia.
When a dangerous defect on chromosome 11q23 disrupts the genetic programming of blood cells, it causes an aggressive and deadly blood cancer called acute myeloid leukemia (AML). With a dismal survival rate of 20-40 percent and desperate need for better treatments, scientists at Cincinnati Children’s report finding a potential therapeutic target for AML in preclinical laboratory tests on donated human cells and mice.
When scientists blocked the target molecule on human AML cells in combination with other known AML treatments, the cancerous blood cells died and were replaced by regenerating, healthy white blood cells, according to principal investigator H. Leighton Grimes, PhD, and study first author Sara Meyer, PhD, a former member of the Grimes laboratory.
The target molecule is F-box protein S-phase kinase-associated protein 2 (Skp2). Skp2 degrades another protein called p27Kip1 that is important to the formation of healthy blood cells.
Published online in the Journal of Experimental Medicine, the study’s findings are not ready for clinical application, according to Grimes. They do suggest the possible development of effective targeted therapies for AML.
“Our work provides a complete mechanistic look into the function of genetic and molecular programs driving this leukemia, and it exploits these processes to identify actionable therapeutic targets,’’ said Grimes, director of the Cancer Pathology Program at Cincinnati Children’s. “We still have extensive additional testing to conduct in laboratory animal models of AML before knowing if this approach will translate to patient care.”
Dangerous Pairs
Abnormalities on 11q23 cause the fusion of harmful genes in aggressive AML blood cancer—the Mixed-Lineage Leukemia (MLL) gene and a multitude of other genetic partners. In this study the investigators closely analyzed MLL-AF9, which includes the AF9 gene, a frequent partner in AML.
One reason AML is so hard to treat is it aggressively re-emerges after initial therapy, which appears at first to diminish the disease in blood cells. But AML is refueled by so-called leukemia stem cells (LSCs)—precancerous blood cells that wait in the wings to evade treatment before launching a full-blown recurrence of disease.
Researchers report they were able to identify and disable the genetic and molecular programming that transform LSCs into AML. But getting to this point required extensive molecular detective work.
Clues in Patient Cells
Extensive biochemical analysis of AML cells donated by patients gave the researchers comprehensive information about the targets and functions of the miR-196 molecular signaling pathway in AML, which the authors said had largely been unknown. The miR-196 microRNA includes a family of microRNAs that help regulate other molecular targets in the cell nucleus. Those other targets depend on the type of cell in which the microRNA is functioning, according to Grimes.
The researchers inserted mimics of the microRNA miR-196 into MLL-AF9 leukemia cells to incorporate them into the cellular machinery. Mimics are strands RNA molecules designed to imitate natural miRNA molecules. Researchers then lysed (disassembled) the cells for analyses, which identified molecular targets of miR-196 in the leukemia cells.
After identifying miR-196 targets, the researchers genetically screened for miR-196 targets in AML cells in mice. These experiments concluded that certain microRNA targets are more important than others in the maintenance and spread of the LSCs that transform into malignant disease.
Additional testing on the cells included computer-assisted analysis of the Molecular Signature Database (a shared multi-institutional research resource). This allowed the researchers to identify sets of genes that show up in high numbers in MLL-AF9 leukemia. This was followed up by additional biochemical testing on the cells. The tests revealed that miR-196 directly targets and inhibits a gene called Cdkn1b/p27Kip1 (cyclin-Dependent Kinase Inhibitor), which controls molecular programming in leukemia stem cell that allows them to maintain aggressive MLL-AF9 leukemia.
Grimes explained that normal blood stem cells (hematopoietic stem cells) are not able to withstand extensive cell division. The researchers discovered that when miR-196 targets Cdkn1b/p27Kip1, it accelerates MLL-AF9 progression by abnormally linking stem cell activity with the growth of leukemia cells.
Killing MLL-AF9 AML Cells
With the data suggesting elevation of p27Kip1 protein levels may be therapeutic to AML patients, researchers investigated a related molecular pathway in the cells that also regulates p27Kip1. This pathway yielded the eventual treatment target Skp2, which degrades the p27 protein and lowers its expression.
When the scientists used an experimental small molecule called SLZ P1041 that inhibits Skp2 on different human AML cell lines, it killed AML depending on the dosage used. Researchers write in their study that the approach “represents a new opportunity for AML therapeutics.”
The researchers also tested SLZ P1041 in combination treatment with other molecule inhibitors (IBET-151, Palbociclib and MI-1) to see if they could get a synergistic therapeutic effect on the AML cell lines. The preclinical data conclude the most consistent synergies were with the combination of SLZ P1041 and an inhibitor of the interaction between Menin and MLL (Menin-MLL) named MI-1.
Funding support for the research came in part from: a postdoctoral fellowship from the Ladies Auxiliary to the Veterans of Foreign Wars; CancerFree KIDS, a University of Cincinnati training grant; the National Institute of Environmental Health Sciences (T32ES007250); a Just-In-Time Core Grant Program award from the Center for Clinical and Translational Science and Training (University of Cincinnati); a Leukemia and Lymphoma Society of America Translational Research Award; the National Institutes of Health (R01CA159845); and The Australian Regenerative Medicine Institute via grants from the State Government of Victoria and the Australian Government.
Contact Information
Jim Feuer
513-636-4656
3333 Burnet Avenue, Cincinnati, Ohio 45229-3026
© 2026 Cincinnati Children's Hospital Medical Center. All rights reserved.


