The Search for a Biological Link between Reactivated HSV and Neurological Disease
The Search for a Biological Link between Reactivated HSV and Neurological Disease
Study Finds HSV Reactivation Kills Neurons and Antivirals Can Stop It
Thursday, March 05, 2020
Reactivated HSV Death Cycle
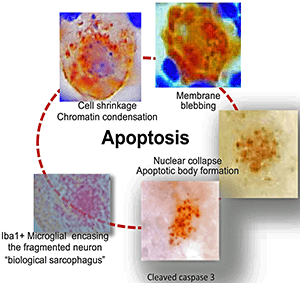
This illustration shows the stepwise apoptotic (a form of cell death) path that a trigeminal sensory neuron undergoes after reactivation of latent HSV. It starts at top left when the chromatin of the neuron undergoing viral reactivation begins to condense, followed by breakdown of the neuronal membrane structure (blebbing), fragmentation and cell death. The neuron cell fragments are removed by Iba1+ macrophage immune cells.
Without even knowing it, most of us carry around latent Herpes Simplex Virus (HSV) in our nervous system—a simple result of being born and living together with others carrying the virus.
New research data in the journal PLoS Pathogens suggests that reactivated HSV in trigeminal nerves of laboratory mice kills off at least a portion of sensory neurons. The findings provide additional evidence that as humans get older, the long-term consequences of HSV infection in the nervous system can accumulate. This can cause neurological damage, report the scientists at Cincinnati Children’s Hospital Medical Center and the University of Cincinnati College of Medicine (UC).
The potential for accrued nervous system damage from HSV reactivation to contribute to neurological diseases like Alzheimer’s is currently a hot topic, according to the researchers. Clinical studies are revealing associations between HSV reactivation and neurodegeneration. Although the correlation between long-term HSV infection and neurodegenerative disease is gaining support, how HSV promotes neurodegeneration isn’t clear.
“We and others are trying to understand the biological consequences of HSV infection in the nervous system over time and how we can develop strategies to prevent these outcomes,” said the study’s principal investigator Nancy Sawtell, PhD, Division of Infectious Diseases at Cincinnati Children’s. “A major question debated by researchers is whether a neuron undergoing viral reactivation survives or dies.”
While the answer to that question is central to understanding HSV pathogenesis, it is not straightforward, she added.
In laboratory studies performed with cultured cells, it appears that specific immune cells control viral reactivation without harming the neuron. However, whether this type of immune control also occurs in the setting of latent virus reactivating in a mouse (or person) remained untested until the current study. Sawtell and her colleagues tested the question by using T cells during the reactivation of latent virus.
Hot Tub Test
Because it isn’t possible to conduct detailed biological studies of HSV reactivation and nerve damage in humans, researchers in this study used laboratory mice that had been given HSV and carried latent viral infections. To stimulate virus reactivation, the mice were subjected to hyper-thermic stress (in a hot tub for laboratory mice). This mimics fever in humans, a known trigger of HSV reactivation.
This allowed the scientists to induce HSV reactivation in the mice and find out what happens to neurons that have undergone HSV reactivation.
“This required painstakingly detailed analysis of the trigeminal ganglia over time,” explained Jessica Doll, PhD, study first author, a post-doctoral fellow at Cincinnati Children’s and former member of the Sawtell lab. “It paid off, allowing us to determine that neurons do not survive viral reactivation but undergo apoptosis, a type of cell death. This is an exciting observation and will form the basis for exciting new research.”
The researchers also studied whether CD4+ and CD8+ T cells played a role in preventing viral reactivation and neuron cell death in mice, but their analysis indicated they do not prevent this outcome.
Proving a Point, Prospecting for Prevention
As part of the effort to find new strategies that might prevent virus reactivation and neuron death, the researchers asked whether amplification of viral DNA in nerves was necessary for reactivation or if the mere expression of viral proteins was sufficient to do this. The availability of antiviral drugs that inhibit amplification of the viral DNA allowed the researchers to test that question.
They used an antiviral drug called acyclovir, which blocks viral DNA amplification. They administered the drug to mouse models that were subjected to hyper-thermic stress to mimic fever. Study data show this prevented HSV reactivation and the death of neurons.
Although further studies are required, Sawtell said this finding holds promise that current antivirals may impact the outcomes of long-term HSV latency and reactivation in the nervous system.
Funding support for the work came in part from: NASA, (NNX13A047G) and the National Institutes of Health (R01 AI093614). Additional support came from the Good Venture Foundation.
Contact Information
Nick Miller
513-803-6035
nicholas.miller@cchmc.org
3333 Burnet Avenue, Cincinnati, Ohio 45229-3026
© 2026 Cincinnati Children's Hospital Medical Center. All rights reserved.


