Rac GTPases, RBC Cytoskeleton and Reactive Oxygen Species in Sickle-Cell Disease
We have demonstrated that deficiency of Rac1 and Rac2 GTPases in mice disrupts the normal hexagonal organization of the RBC cytoskeleton and reduces erythrocyte deformability. This is associated with increased phosphorylation of adducin at Ser-724, (corresponding to Ser-726 in human erythrocytes), a domain target of protein kinase C (PKC). PKC phosphorylates adducin and leads to decreased F-actin capping and dissociation of spectrin from actin, implicating a significant role of such phosphorylation in cytoskeletal remodeling. We evaluated adducin phosphorylation in erythrocytes from patients with sickle-cell disease and found it consistently increased at Ser-726. In addition, ROS concentration is elevated in sickle erythrocytes by 150-250 percent compared to erythrocytes from normal control individuals.
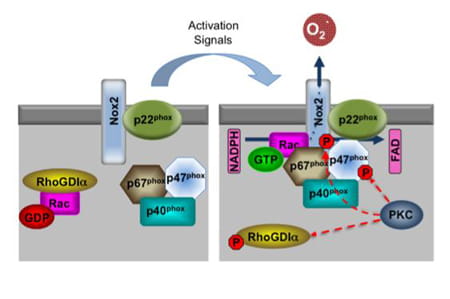
Rac1 and Rac2 GTPases mediate reactive oxygen species (ROS) production via NADPH oxidase in a variety of cells. Inactive GDP-bound Rac proteins are sequestered in the cytosol by Rho GDP-dissociation inhibitors (RhoGDIs), which bind and mask the hydrophobic C-terminal region of Rac, the same region that is responsible for targeting Rac to the plasma membrane. The Rac GTPases are a critical component of NADPH oxidase complex, and their recruitment from the cytoplasm into the membrane-bound complex is necessary for NADPH oxidase activity.
The phagocyte NADPH oxidase was the first identified and best-studied member of the NOX family, contributing to microbial killing. Nox2 (previously called gp91phox) along with the p22phox subunit localizes in the membrane of intracellular vesicles in resting neutrophils. Upon activation, Rac GTPases mediate the association of p67phox with the catalytic subunit Nox2 to form the NADPH oxidase complex within the plasma or phagosomal membrane, which functions as a transmembrane redox chain that connects the electron donor (NADPH) in the cytosol with the electron acceptor (molecular O2) on the other side of the membrane (extracellular or intravesicular) to produce superoxide radicals.
Several studies provide evidence that PKC activation induces NADPH oxidase-mediated ROS production. In aortic endothelial and renal mesangial cells, it has been shown that high-glucose levels stimulate ROS production via a PKC-dependent activation of Rac1 and NADPH oxidase, and that this activation is inhibited by PKC inhibitors. In epithelial cell lines, activation of PKC results in increased phosphorylation of RhoGDIα that dissociates to allow Rac to translocate to the membrane and interact with membrane-associated activators. In neutrophils, PKC has been shown to activate NADPH oxidase by phosphorylation of Nox2 or phosphorylation of p47phox. Whether a similar mechanism is at work to generate ROS in erythroid cells, and more intriguingly, if abnormality of such a ROS-producing molecular complex is associated with sickle-cell disease phenotype, awaits future investigation (George et al. Blood Cells, Molecules, and Diseases. 45:41–45. 2010).