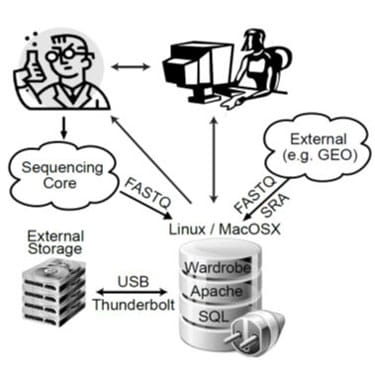
The recent proliferation of next-generation sequencing (NGS)–based methods for analysis of gene expression, chromatin structure and protein-DNA interactions has opened new horizons for molecular biology. These methods include RNA sequencing (RNA-Seq), chromatin immunoprecipitation sequencing (ChIP-Seq), DNase I sequencing (DNase-Seq), micrococcal nuclease sequencing (MNase-Seq), assay for transposase-accessible chromatin sequencing (ATAC-Seq) and others. On the “wet lab” side, these methods are largely well established and can be performed by experienced molecular biologists; however, analysis of the sequenced data requires bioinformatics expertise that many molecular biologists do not possess. Utilizing published datasets is also challenging: although authors usually comply with the longstanding requirement to deposit raw data files into databases such as Sequence Read Archive (SRA) or Gene Expression Omnibus (GEO), it is impossible to analyze these datasets without special expertise. Even when processed data files (e.g., gene expression values) are available, direct comparison between datasets is ill advised because different laboratories use different pipelines (or different software versions). This situation means that biologists require the help of bioinformaticians even for the simplest of tasks, such as viewing their own data on a genome browser, putting these exciting techniques beyond the reach of many laboratories. Even when bioinformaticians are available, differences in priorities within collaborations can result in delays and misunderstandings that are damaging to the research effort. An optimal way to mitigate these problems is to enable biologists to perform at least basic tasks without the help of bioinformaticians by creating user friendly data analysis software.
We therefore developed BioWardrobe—a biologist-friendly platform for integrated acquisition, storage, display and analysis of NGS data, aimed primarily at researchers in the epigenomics field. BioWardrobe’s features include download of raw data from core facilities or online databases (e.g., GEO), read mapping and data display on a local instance of the UCSC genome browser, quality control and both basic and advanced data analysis. In basic analysis, automated pipelines are used to process each experiment. The pipelines are selected on the basis of biologist-friendly experimental parameters (e.g., RNA/ChIP-Seq, paired or single, genome, stranded or unstranded, antibody) and combine the tools developed by ourselves and by others (e.g., Bowtie, STAR, FASTX and MACS2) with wrappers that enhance the output of original software by offering additional information (e.g., assigning ChIP/DNase-Seq peaks to the nearest genes), provide experimentally meaningful quality controls and display results within the web interface. The quality controls produced during basic analysis were chosen to facilitate troubleshooting of experimental procedures. Customizable advanced analysis can combine multiple experiments and includes tools for comparing gene expression (DESeq1/2) and genome occupancy (MAnorm28) profiles between samples or groups of samples and creating principal component analysis (PCA) plots, gene lists, average tag density profiles and heatmaps using a graphical user interface. Incorporating additional custom scripts is facilitated by a built-in interface for the R programming language. All the precomputed data are stored in an SQL database and can be accessed via a convenient web interface by biologists. Bioinformaticians, on the other hand, can access the data from R using a provided R library or using other programming languages with standard MySQL libraries. BioWardrobe can be run on Linux or MacOSX systems (e.g., a Mac Pro desktop). The installation package and instructions are available at the BioWardrobe website and via the GitHub website under GNU GPL v2.
Development of BioWardrobe has led to many successful collaborations with researchers both within and outside Cincinnati Children’s. We have now converted the BioWardrobe server into a core facility that provides data analysis service to more than 30 laboratories at Cincinnati Children’s. We are also continuing development of BioWardrobe by integrating novel tools and switching our pipelines to Common Workflow Language.

 Modern biomedical research has seen a remarkable increase in the production and computational analysis of large datasets, leading to an urgent need to share analytical techniques. The evolving complexity of computational tools and pipelines makes it nearly impossible to reproduce computationally heavy studies or to repurpose published analytical workflows. Even when the tools are published, the lack of description of operating system environments or software component versions can lead to either unfaithful reproduction of the analysis or analysis failing altogether when performed within a differing environment. In order to ameliorate this situation, a team of researchers and software developers has formed a
Modern biomedical research has seen a remarkable increase in the production and computational analysis of large datasets, leading to an urgent need to share analytical techniques. The evolving complexity of computational tools and pipelines makes it nearly impossible to reproduce computationally heavy studies or to repurpose published analytical workflows. Even when the tools are published, the lack of description of operating system environments or software component versions can lead to either unfaithful reproduction of the analysis or analysis failing altogether when performed within a differing environment. In order to ameliorate this situation, a team of researchers and software developers has formed a 


