Improving Child Health Through Basic, Clinical and Translational Research
The Division of Critical Care Medicine engages in basic, clinical and translational research spanning the spectrum of pediatric critical illness. Our researchers are dedicated to improving child health, whether they are studying functional mechanisms of disease, focusing on prevention, or developing novel diagnostic, therapeutic or care delivery strategies.
Our Research
Critical care medicine faculty at Cincinnati Children’s work side-by-side to develop new research initiatives, design studies, interpret data and apply their discoveries to the patient care setting. The division’s comprehensive research programs are funded through grants from the National Institutes of Health, the Cincinnati Children’s Research Foundation, and support from other research organizations and industry partners. Our researchers publish in high-profile medical journals, deliver presentations at medical conferences and train the next generation of clinical, academic and research leaders in pediatric critical care.
Understanding and Preventing Sepsis
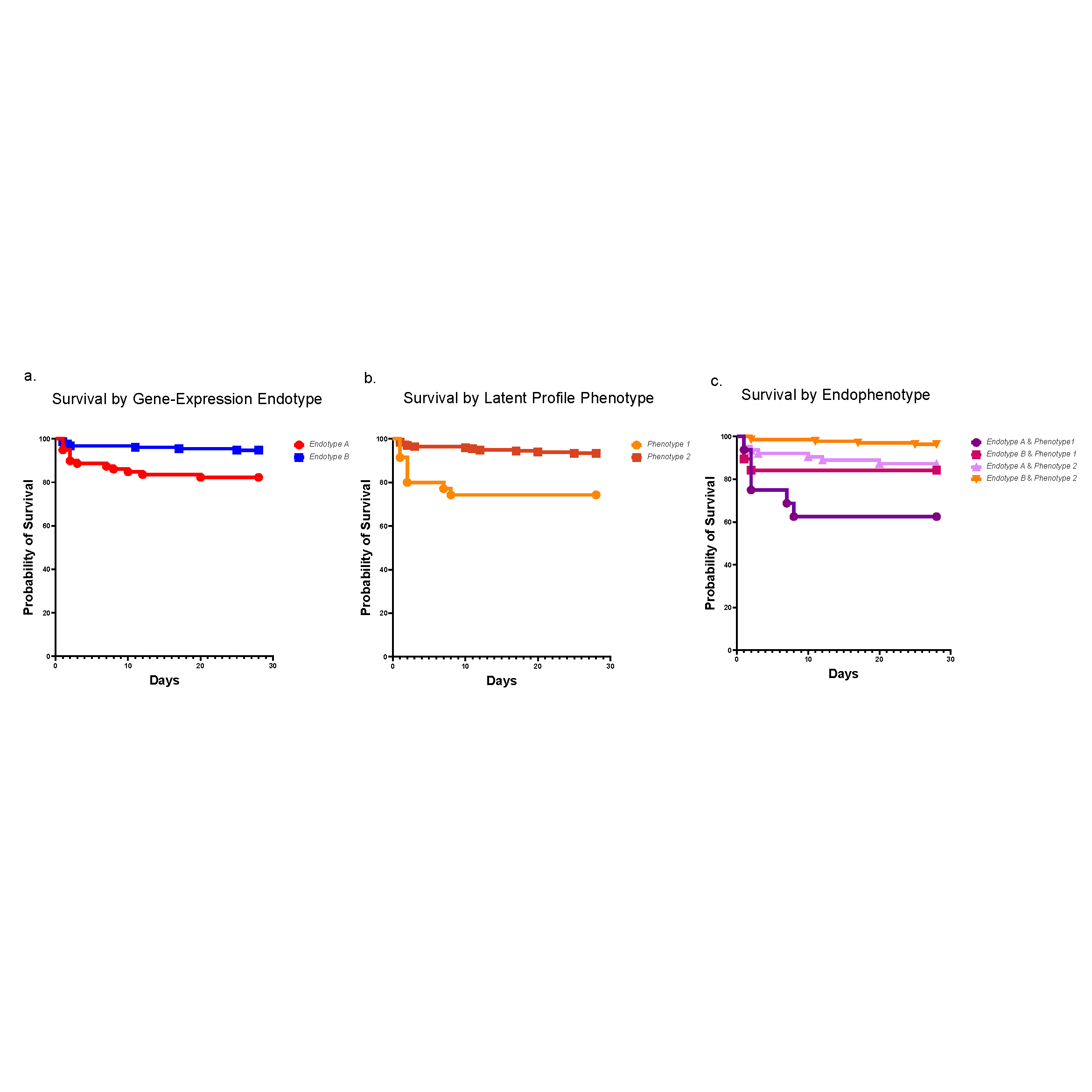
A historical and ongoing focus of our division’s research is sepsis, a disease that kills 11 million people each year, many of them children. In recent years, researchers at Cincinnati Children’s have made significant contributions to the field of sepsis care—spanning from defining cellular mechanisms of organ injury to risk stratification, early recognition and optimization of care delivery. One example is our leadership role in developing the Pediatric Sepsis Biomarker Risk Model (PERSEVERE). This biomarker-based stratification tool uses genome-wide expression profiling to estimate a specific patient’s baseline mortality risk from sepsis.
Other areas of focus include:
- Partnering with other pediatric centers through PALISI, the Pediatric Lung Injury and Sepsis Investigator Network, to advance scientific discovery through clinical research
- Leading a national effort to stratify patients according to specific inflammatory genotypes to develop precision medicine strategies for various types of multi- and specific-organ failure in children
- Seeking to understand the mitochondrial dysfunction in multiorgan failure in trauma and sepsis, using in vitro and in vivo experimental models
- Engaging with the community to prevent critical illness in children and improve access to care
- Using clinical informatics approaches to optimize the use of the electronic medical record, with the goal of improving clinical decision support in the pediatric intensive care unit
- Using virtual and augmented reality, artificial intelligence and other innovative approaches to enhance training and care delivery